
EN

近日,美国食品药品监督管理局(FDA)发布《合理机制框架用于已知病因特定遗传病个体化治疗开发》(2026草案)。
该指南是美国FDA下属CBER(生物制品评估和研究中心)与CDER(药品评估和研究中心)联合发布的行业草案指南,2026年2月正式公开征求意见,是全球首个系统性规范超罕见遗传病个体化治疗全生命周期开发的监管文件。它首次将 “合理机制框架(Plausible Mechanism Framework)” 上升为正式监管路径,彻底解决了因患者数量极少(甚至单例患者)无法开展传统随机对照试验(RCT)的行业痛点,为基因组编辑(GE)、反义寡核苷酸(ASO)等个体化疗法的快速获批提供了明确依据。
适用对象:针对已知明确生物学病因的严重致残 / 致死性遗传病(SDLT),且因靶点特异性和患者极端罕见性无法开展传统 RCT 的个体化治疗,重点覆盖基因组编辑产品(CRISPR、碱基编辑、先导编辑等)和 RNA 靶向疗法(ASO、siRNA),其原则也可延伸至其他类型个体化治疗。
监管路径:
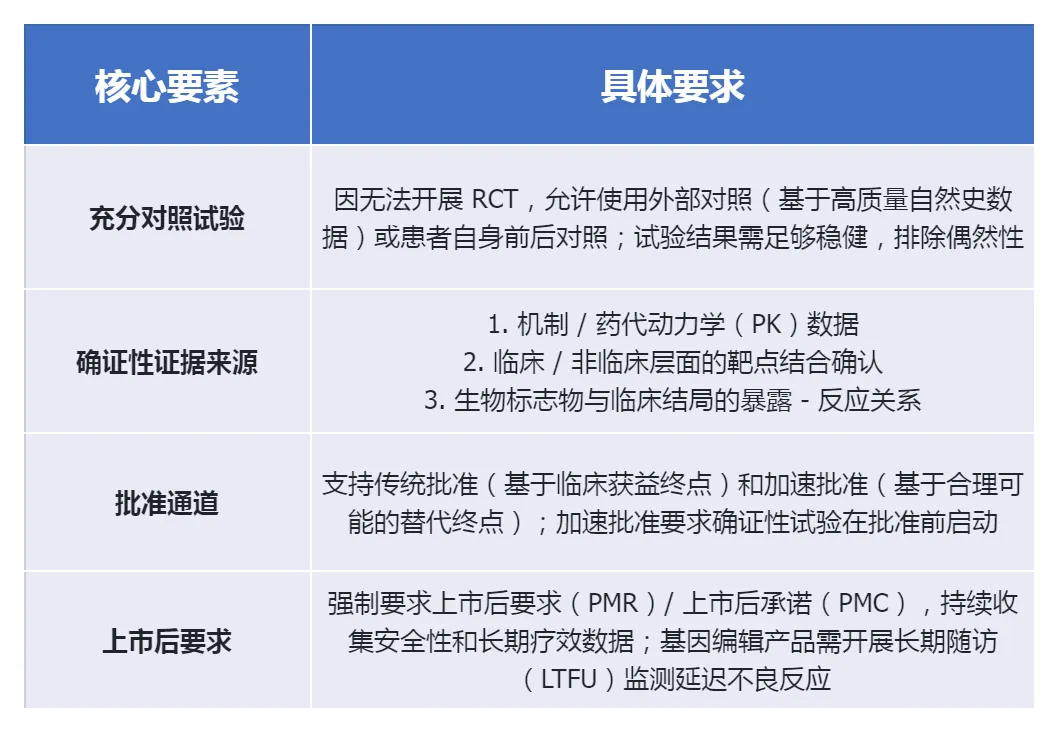
突破性的“单试验+确证证据”模式
本指南最大突破是明确:个体化治疗可通过“1项充分且良好对照的临床试验+确证性证据”满足FDA“实质性有效性证据”要求,无需开展多项RCT。
非临床研究:
高度灵活性 + 数据杠杆化
非临床研究的核心目标是证明概念验证(POC)和支持首次人体试验(FIH)的安全性,而非完成传统全套毒理试验,FDA给予了极大的监管弹性。
1. 通用原则
鼓励使用新方法学(NAMs):包括患者来源细胞、类器官、计算机模拟、化学模拟等,替代部分动物实验。
允许混合研究设计:可将POC、安全性、生物分布研究合并为一项“确定性混合研究”,大幅缩短开发周期。
强制要求早期沟通:建议在IND提交前与FDA讨论非临床开发计划,特别是数据杠杆化策略。
2. 基因组编辑(GE)产品专项要求
POC研究:必须使用NGS方法定量评估靶向编辑效率;可使用物种同源编辑产品,但需提供生物学相关性证明。
安全性评估:全面覆盖载体毒性、编辑组件毒性、免疫原性、脱靶编辑(NGS检测低频事件)和染色体完整性;体内产品需评估生物分布,体外产品需评估细胞存活 / 植入。
数据杠杆化:同一编辑器平台的不同gRNA变体可共享核心非临床数据,仅需补充每个变体的靶向活性和脱靶风险评估。
3. ASO产品专项要求
成熟化学类ASO:可采用简化非临床方案,仅需核心安全药理学、标准毒理学和剂量探索研究;可引用公开的同类产品毒性数据。
非成熟化学类ASO:需开展更全面的传统非临床研究。
脱靶评估:必须采用生物信息学+体外实验方法评估杂交依赖的脱靶结合风险。
CMC:
平台化数据共享,加速工艺验证
针对个体化治疗批次少、开发周期短的特点,FDA允许最大化利用平台知识和历史数据,简化CMC要求。
IND阶段:用于Ⅰ期临床试验的产品可豁免21 CFR Part 211的部分CGMP要求。
NDA/BLA 阶段:
必须拥有验证的生产工艺,但同一平台的不同产品变体可共享工艺验证数据。
分析方法:已验证的同类产品方法可通过 “适用性评估” 直接使用;效价测定需设计为可适配不同突变变体。
稳定性:因批次数量有限,允许利用同类产品数据支持有效期设定,需采用稳定性指示性检测方法。
模块化GE产品:仅更换gRNA的产品变体,可共享大部分CMC信息,仅需补充变体特异性的质量控制数据。
原文附件
