
中

Recently, the China Biotechnology Development Center released the "Guidelines for the Clinical Research of New Technologies for Induced Pluripotent Stem Cells (Version 1)", which will come into effect on May 1, 2026. It is a technical document in support of the "Regulations on Clinical Research and Clinical Transformation and Application of Biomedical New Technologies" issued by the State Council (Order No. 818).
As the first national technical guideline specifically targeting clinical research on pluripotent stem cells in China, this guideline has filled the gap in the previous regulatory field of pluripotent stem cells. It marks that the treatment of pluripotent stem cells in China has officially entered a new stage of standardization and normalization from the previous relatively loose "exploratory research" phase.
This guideline is applicable to clinical research on new technologies for pluripotent stem cell therapy conducted within China, which is not aimed at drug registration. It specifically covers treatment technologies derived from human induced pluripotent stem cells (hiPSCs), human embryonic stem cells (hESCs), gene-modified pluripotent stem cell treatment technologies, and functional cell treatment technologies obtained through pluripotent stem cell induction and differentiation.
At the same time, the guidelines clearly exclude four types of ineligible situations: clinical trials conducted for the purpose of drug registration (such trials should be applied for with the National Drug Administration); hematopoietic stem cell transplantation technology; reproductive cell/embryo gene editing technology; and autologous cell reinfusion technology without in vitro manipulation.
Core filing requirements
1. Institutional and personnel requirements (high entry standards)
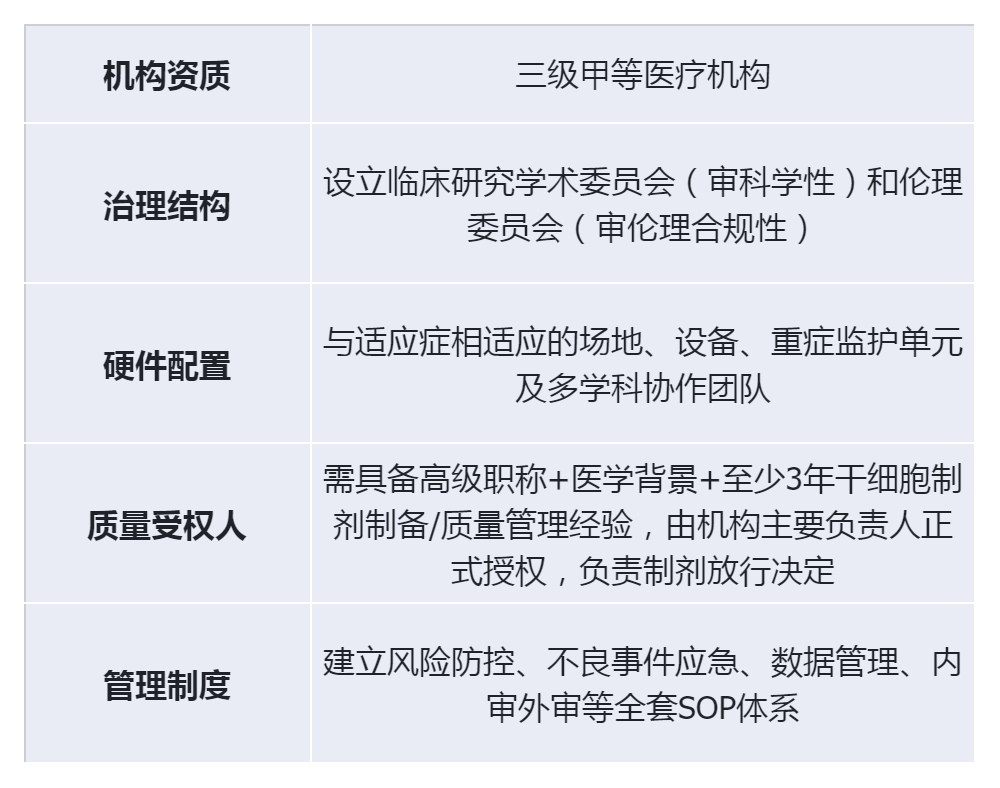
2. Institutional and personnel requirements (high entry standards)
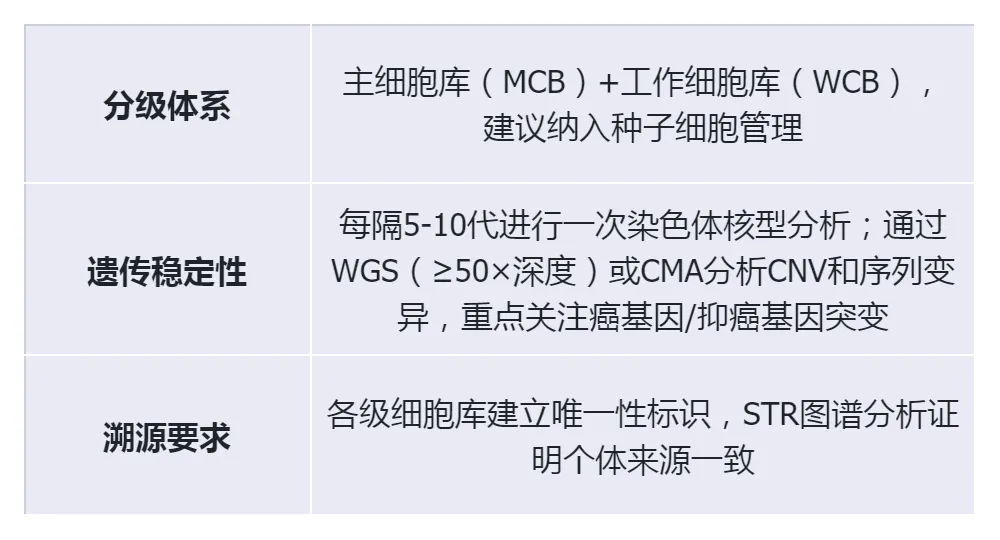
(1)Cell bank management
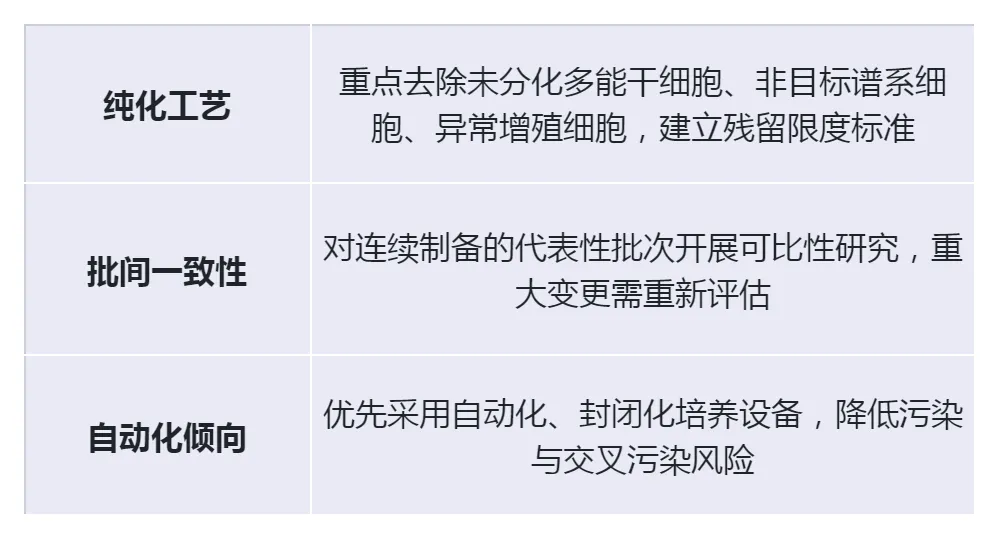
(2)工艺控制
(3)Core indicators of quality control (core safety indicators)

(4)Special requirements for genetic modification
Based on the different stages of the process (before database establishment vs. during differentiation), formulate differentiated control strategies (for benefit-risk assessment)
The lentiviral vector needs to be tested for replicative lentivirus (RCL), and the γ-reverse transcriptase virus needs to be tested for replicative reverse transcriptase virus (RCR).
Evaluation of off-target editing, insertion mutations and genetic stability of the target gene
3. Non-clinical research (benefit-risk assessment)
The four key points of safety assessment:
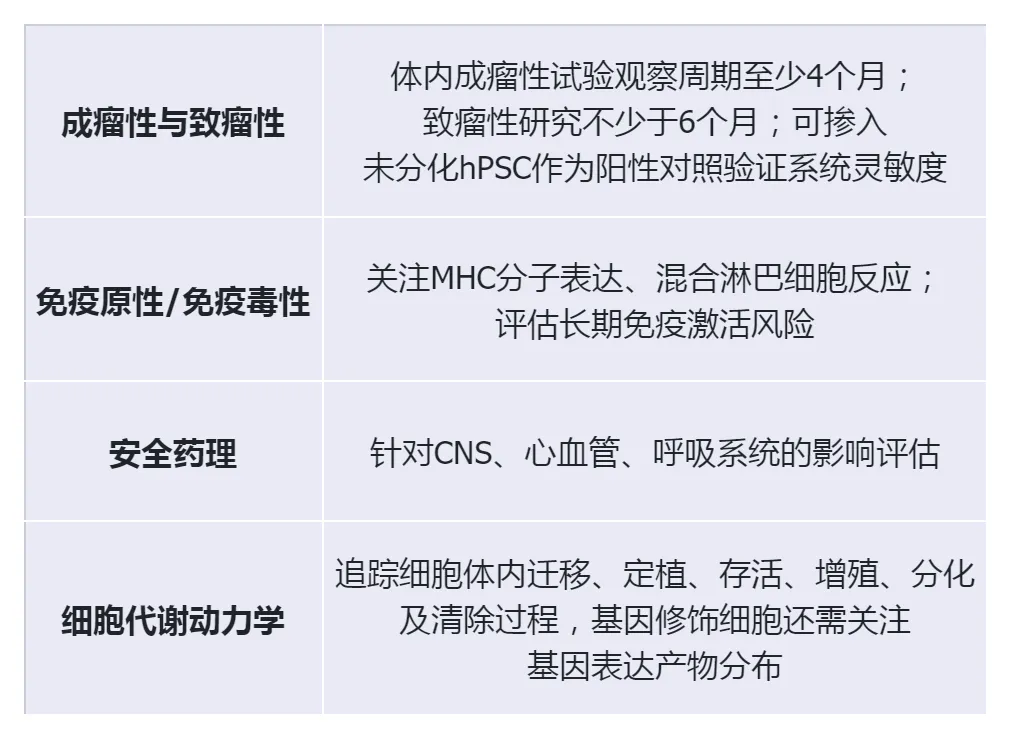
Evaluation of effectiveness:
At least obtain valid data on the effectiveness of one relevant animal species or model
When animal models are lacking, organoids and microfluidic models can serve as supplementary evidence.
4. Clinical research design (risk stratification management)
(1)Research Type
Exploratory study: dose escalation, sentinel dosing, batch enrollment, prioritizing the inclusion of subjects with high homogeneity and few confounding factors
Confirmatory study: Preferentially adopt random, controlled, and double-blind design, and ensure the representativeness of the sample size.
(2)Subject protection
Children/Adolescents: Generally initiated after adults have obtained preliminary safety data; additional attention should be paid to effects on growth and development, neurocognition, and reproductive system.
Exit/termination mechanism: The plan must clearly define the management principles and handling procedures for situations where participants withdraw midway, the study is suspended or terminated, including measures for stopping the infusion, conducting safety assessments, providing medical treatment and referral, and conducting follow-up.
(3)Long-term follow-up (differential duration)
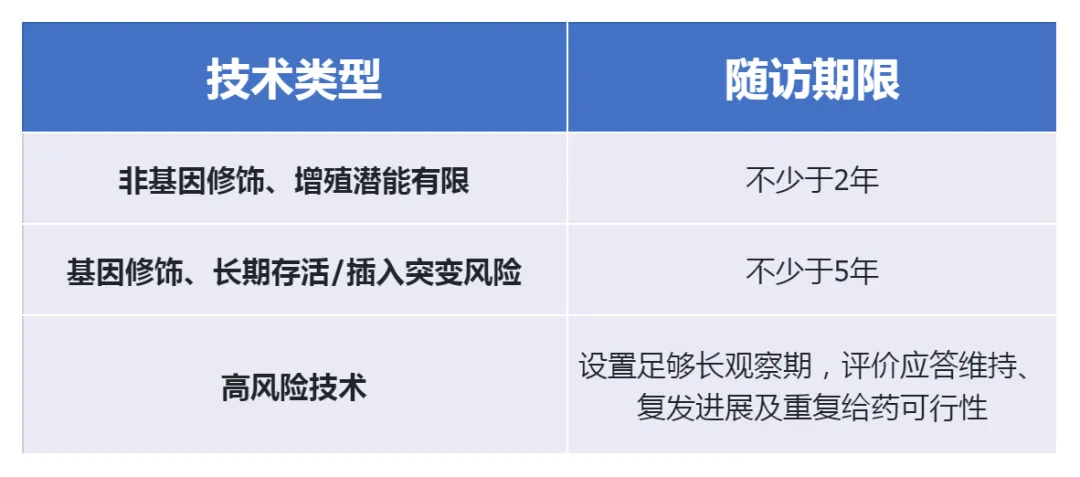
(4)Formulation Release and Traceability
Clinical research institutions must conduct re-inspections and evaluations on the preparations transported to the hospital. Those that fail the inspection shall not be used.
The multi-center study was conducted under a unified plan, SOPs, training, and the centralized reporting of adverse events by the main institution.
5.Special requirements for ethical compliance
(1)In addition to the regular requirements, ethical review should also focus on the following aspects of assessment:
Ethics of Cell Sources:
Source of the sample: Review of the ethical approval for the collection of the original sample, donor screening, and the scope of informed consent authorization.
Source of human embryos: Confirm the use of only the remaining embryos from IVF procedures and those voluntarily donated within 14 days.
Self-source: Review of collection methods, risk management of cross-contamination, and protection of personal information
(2)Special Risk Benefit Assessment:
Classify and grade the assessment of tumor-forming/carcinogenic properties, ectopic cell growth, long-term immune activation, off-target effects of gene modification, and the risk of insertional carcinogenesis.
Thoroughly review the non-clinical research support, formulation quality control, process consistency and traceability
Attached file
