
中

Recently, the US Food and Drug Administration (FDA) released "Framework for Reasonable Mechanisms for the Development of Individualized Therapies for Specific Genetic Diseases with Known Causes" (draft 2026).
This guideline is an industry draft guideline jointly released by CBER (Biological Products Evaluation and Research Center) and CDER (Drug Evaluation and Research Center) under the US FDA. It was officially open for public comment in February 2026 and is the world's first regulatory document that systematically standardizes the full life cycle development of individualized treatment for ultra-rare genetic diseases. It has elevated the "Plausible Mechanism Framework" to a formal regulatory path, completely resolving the industry pain point of being unable to conduct traditional randomized controlled trials (RCT) due to the extremely small number of patients (even a single patient), and providing a clear basis for the rapid approval of individualized therapies such as genome editing (GE) and antisense oligonucleotides (ASO).
Applicable Subjects: For severe disabling/lethal genetic diseases with a clearly identified biological cause (SDLT), where individualized treatment cannot be carried out using traditional RCT due to the specificity of the target and the extreme rarity of the patients. The focus is on genomic editing products (CRISPR, base editing, lead editing, etc.) and RNA-targeted therapies (ASO, siRNA). The principle can also be extended to other types of individualized treatments.
Regulatory path:
The groundbreaking "single trial + confirmatory evidence" model
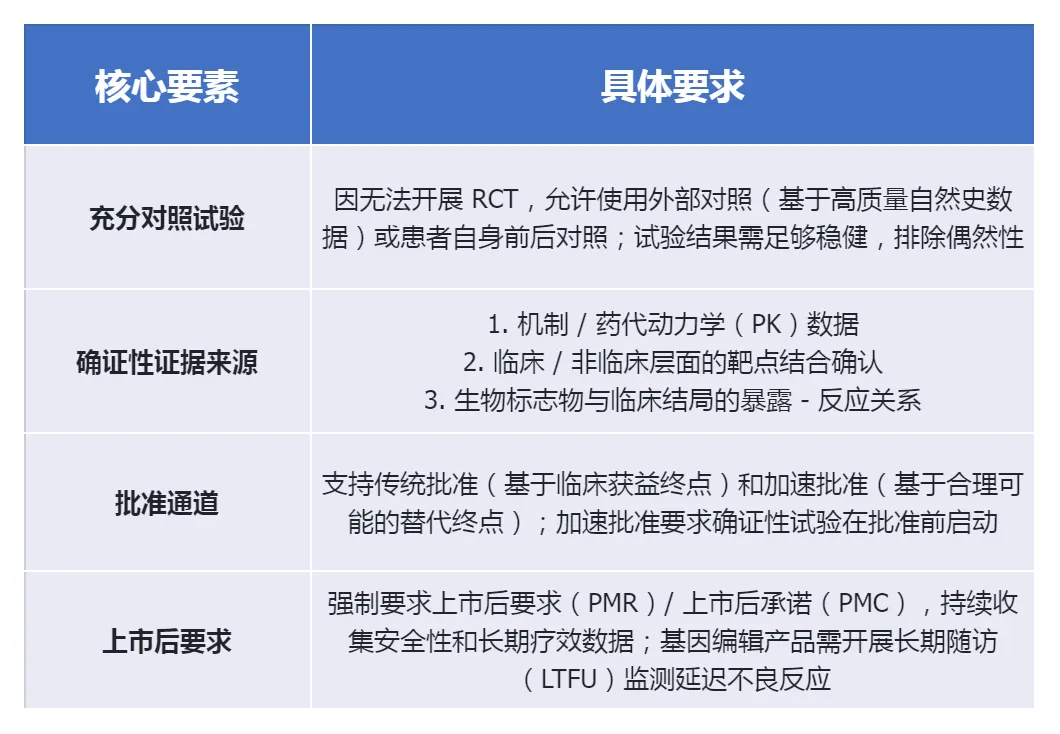
The most significant breakthrough of this guideline is that it clearly states that individualized treatment can meet the FDA's requirement for "substantial evidence of effectiveness" through "one fully and well-controlled clinical trial plus confirmatory evidence", without the need to conduct multiple RCTs.
Non-clinical research:
High flexibility + Data leverage
The core objective of non-clinical research is to demonstrate proof of concept (POC) and to ensure the safety of the first-in-human trial (FIH), rather than completing the full range of traditional toxicology tests. The FDA has granted significant regulatory flexibility in this regard.
1. General principle
Encourage the use of new methodologies (NAMs): such as patient-derived cells, organoids, computer simulations, chemical simulations, etc., to replace some animal experiments.
Allow for mixed research designs: The POC, safety, and biodistribution studies can be combined into a "deterministic mixed study", significantly shortening the development cycle.
It is mandatory to conduct early communication: It is recommended to discuss the non-clinical development plan, especially the data leveraging strategy, with the FDA before submitting the IND.
2. Special requirements for genome editing (GE) products
POC study: The targeted editing efficiency must be quantitatively evaluated using NGS methods; species homologous editing products can be used, but a proof of biological relevance must be provided.
Safety assessment: Comprehensive coverage of vector toxicity, editing component toxicity, immunogenicity, off-target editing (NGS detection of rare events), and chromosome integrity; in vivo products need to be evaluated for biological distribution, and in vitro products need to be evaluated for cell survival/implantation.
Data leveraging: Different gRNA variants on the same editor platform can share the core non-clinical data, with only the assessment of the targeting activity and off-target risks for each variant needing to be supplemented.
3. ASO Product Specific Requirements
Mature chemical ASO: A simplified non-clinical protocol can be adopted, requiring only core safety pharmacology, standard toxicology, and dose exploration studies; publicly available toxicity data of similar products can be cited.
Non-mature chemical-based ASO: More comprehensive traditional non-clinical studies need to be conducted.
Off-target assessment: The risk of hybrid-dependent off-target binding must be evaluated using a combination of bioinformatics and in vitro experimental methods.
CMC:
Platform-based data sharing accelerates process validation
Given the characteristics of individualized treatment involving a small number of batches and a short development cycle, the FDA allows for the maximum utilization of platform knowledge and historical data to simplify CMC requirements.
IND stage: Products for Phase I clinical trials are exempt from some of the CGMP requirements stipulated in 21 CFR Part 211.
NDA/BLA stage:
It is necessary to have verified production processes, but different product variants on the same platform can share the process verification data.
Analysis method: The validated methods for similar products can be directly used through "applicability assessment"; the potency determination needs to be designed to be adaptable to different mutant variants.
Stability: Due to the limited batch quantity, it is allowed to use data from similar products to support the determination of the validity period. Indicative stability testing methods should be adopted.
Modular GE products: Product variants that only require the replacement of gRNA can share most of the CMC information, and only need to supplement the quality control data specific to the variants.
Attached file
