
中

Source: BioArt
Alzheimer's Disease (AD) is one of the most common neurodegenerative diseases in the elderly population, its pathogenesis has not been fully clarified, and there is no complete cure. In recent years, genome-wide association studies (GWAS) and related genetic studies have revealed thousands of single nucleotide polymorphic loci (SNPS) associated with Alzheimer's disease. Because the pathogenesis of Alzheimer's disease is extremely complex, most of the associated sites are in the non-coding regions of the genome, which sites will increase the risk of Alzheimer's disease, and its specific effects and mechanisms need to be further studied.
There are a large number of candidate cis-regulatory elements (cCRE) with potential gene expression regulation functions in the non-coding regions of the genome, such as enhancers, silencers, etc., which can regulate the transcriptional activity of target genes through long-distance interaction with promoters. Previous studies have shown that more than 90% of Alzheimer's disease association sites are concentrated in cCRE sequences, so elucidation of the regulatory functions of these cCRE target genes is the key to elucidate the genetic risk of Alzheimer's disease. As immune cells in the brain, microglia not only play an important role in immune response, monitoring and maintaining immune homeostasis in the brain, but also more associated sites related to Alzheimer's disease are enriched in cCRE sequences of microglia than neurons, astrocytes and oligodendrocytes. It is suggested that microglia are of great significance for the study of cCRE at the associated sites of Alzheimer's disease and the in-depth analysis of its genetic mechanism.
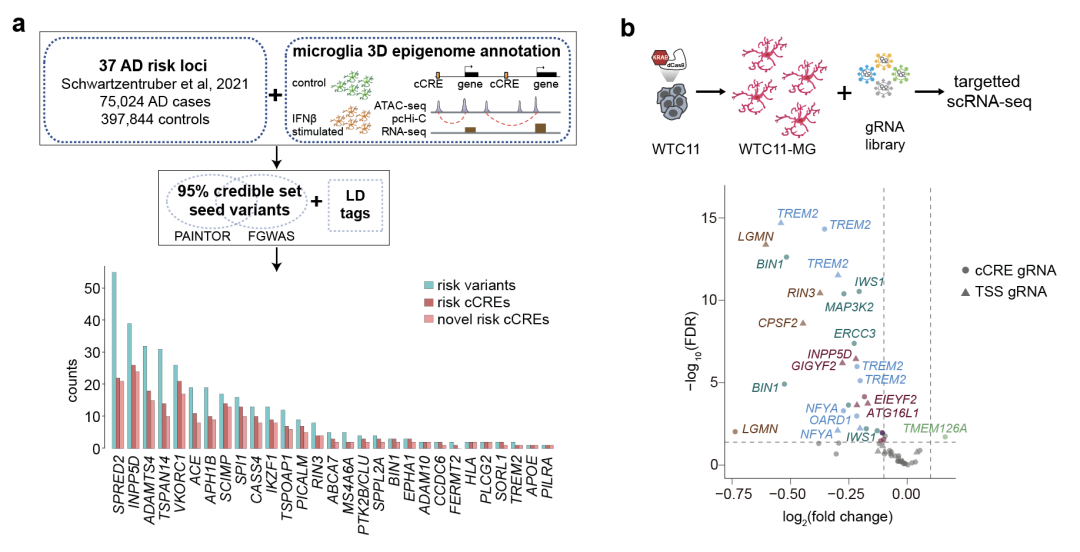
On September 21, 2023, Shen Yin Research Group at the University of California, San Francisco and the University of North Carolina, Chapel Hill Yun Li and his colleagues at Chapel Hill have published a paper in Nature Genetics entitled Functional characterization of Alzheimer's disease genetic variants microglia's paper, for the first time, combined microglia epigenetic information with Alzheimer's GWAS data to systematically identify genetic loci highly associated with Alzheimer's disease, Moreover, single-cell CRISPR interference screening technology was used to explain that multiple Alzheimer's disease associated loci regulate target gene expression through long-distance chromosome interaction and affect the physiological function of microglia, providing a research example for the comprehensive analysis of the genetic mechanism of Alzheimer's disease.

Studies have shown that viral infection can significantly increase the risk of a variety of neurodegenerative diseases [1]. Therefore, We investigated the different transcription profiles (RNA-seq), open chromatin region maps (ATAC-seq), and high-precision 3D chromatin interaction maps of microglia derived from human pluripotent stem cells (hPSC) under resting and interferon IFNβ stimulation (simulated viral infection) (promoter capture Hi-C) to explain the gene expression regulation mechanism of microglia in two states: resting and responding to virus infection. Interestingly, the authors found that the chromatin open state and 3D interactions corresponding to most interferon-stimulated differentially expressed genes remained relatively stable before and after stimulation, suggesting that cells already had most of the prerequisites for rapidly responding to transcriptional regulatory changes in the stimulus. On the other hand, genes with simultaneous changes in chromatin open status and 3D interactions were more likely to have expression changes. For example, a cCRE downstream of the Alzheimer's risk gene MS4A6A contains two genetic loci. After interferon stimulation, this cCRE exhibited a stronger chromatin open state and a stronger 3D interaction with the MS4A6A promoter. These epigenetic phenomena well explain the elevated transcription levels of MS4A6A after interferon stimulation, suggesting that the genetic locus of this cCRE may increase an individual's risk of Alzheimer's disease by regulating immune response genes.
Next, the researchers combined the non-coding genetic loci associated with Alzheimer's disease with the three-dimensional epigenome map of human microglia to reveal 308 new strongly associated Alzheimer's disease-related genetic loci located in 181 CCres. By examining allelic effects on transcription of nearby genes, the authors further prioritized 118 loci from 25 loci. The team's study of these priority sites and the function of cCREs revealed a range of genetic mechanisms associated with Alzheimer's disease. For example, through single-cell CRISPR interference screening experiments in human microglia, the authors observed that a single Alzheimer's disease-associated functional site can affect the expression of multiple genes, and that the combined action of multiple cCREs can have a nonlinear synergistic effect on the expression of target genes. These results highlight the complexity of Alzheimer's disease risk driven by non-coding region-mediated transcriptional regulation and underscore the importance of studying the combined effects of cCREs in gene regulation.
In addition, the research team also used prime editing technology [2] to perform gene editing and allele expression analysis on two loci in the same cCRE in the intron region of risk gene TSPAN14, and determined that rs7922621 directed regulation of TSPAN14 gene expression. The membrane transport of ADAM10 and the shear process of soluble TREM2 were further affected. Given that TSPAN14, ADAM10, and TREM2 are all Alzheimer's risk genes, this study enables a systematic assessment of the mechanisms of action of Alzheimer's disease-associated variants in microglia, covering the full spectrum from genetic fine-prediction to linking genetic association sites to disease-associated molecular and cellular functions.
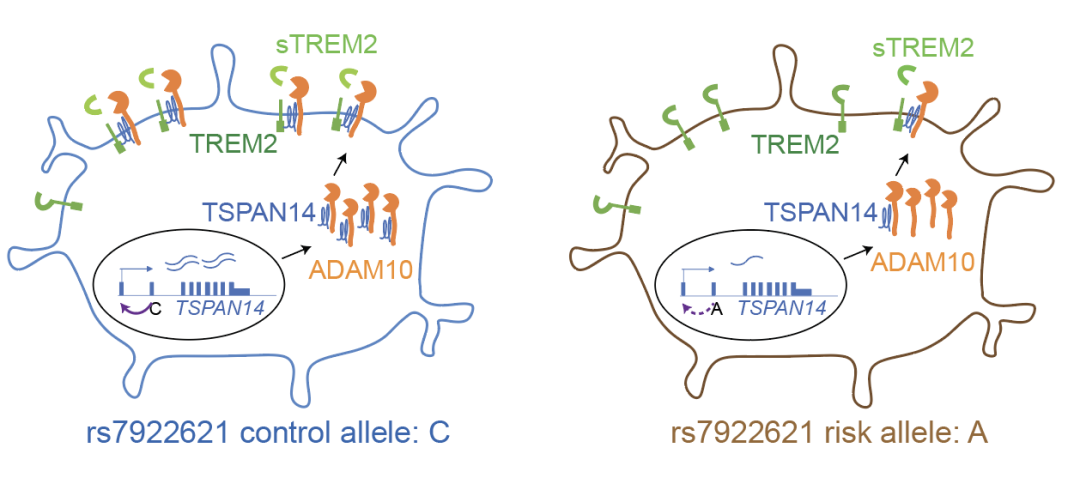
It is worth emphasizing that the results of TSPAN14 site in this study showed that rs7922621 in the same cCRE and its completely positive correlation site rs7910643 did not show the above molecular or cellular level functions, highlighting the importance of the single-base level of lead editing for accurate genomic function analysis. There are often multiple genetic loci associated with complex diseases in the same cCRE, and it is difficult to pinpoint disease-causing sites precisely because common gene-editing methods, such as CRISPR, are difficult to achieve single-base accuracy. Shen Yin's research group recently developed PRIME (High-throughput single-base in situ Editing Screening) method, which realized functional screening of human genome with single-base resolution and high throughput [3]. This technology will significantly improve our ability to elucidate the genetic mechanisms of complex diseases, improve early diagnosis and risk assessment, develop new drugs, and develop personalized treatments.
The study was also supported by Professor Li Gan's team from the Helen and Robert Appel Alzheimer's Disease Research Institute/Weill Cornell Medical College (Helen and Robert Appel Alzheimer's Disease Research Institute, Weill Cornell Medical College). Dr. Xiaoyu Yang of Shen Yin's research group is the first author of the article.
Original link:https://www.nature.com/articles/s41588-023-01506-8
References:
1.Levine, K. S. et al. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron, doi:10.1016/j.neuron.2022.12.029 (2023).
2.Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149-157, doi:10.1038/s41586-019-1711-4 (2019).
3.Ren, X. et al. High throughput PRIME editing screens identify functional DNA variants in the human genome. bioRxiv, 2023.2007. 2012.548736 (2023).